

The laboratory’s research program encompasses five coordinated research foci.
A Dynamic Signaling Code to specify cellular stress responses
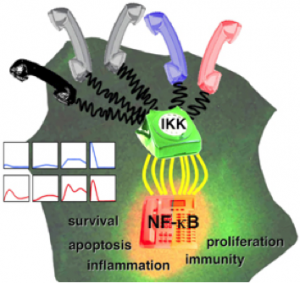 Using the IκB-NFκB signaling module as a model system, we have studied the mechanisms that encode NFκB dynamic control and uncovered multiple parallel negative feedback mechanisms (IκBα, IκBβ, IκBε) that operate at different timescales and thus can have stimulus-specific effects (25, 31, 37, 43, 57, 62, 81). We have also applied the systems biology approach to study the mechanisms (A20 and autocrine TNF) that encode stimulus-specific IKK dynamics (37, 57). We have recently made progress in understanding the control of specificity of the multifunctional IKK complex, via for example the scaffold functions of NEMO (85).
Using the IκB-NFκB signaling module as a model system, we have studied the mechanisms that encode NFκB dynamic control and uncovered multiple parallel negative feedback mechanisms (IκBα, IκBβ, IκBε) that operate at different timescales and thus can have stimulus-specific effects (25, 31, 37, 43, 57, 62, 81). We have also applied the systems biology approach to study the mechanisms (A20 and autocrine TNF) that encode stimulus-specific IKK dynamics (37, 57). We have recently made progress in understanding the control of specificity of the multifunctional IKK complex, via for example the scaffold functions of NEMO (85).
These studies have had impact on the field, influencing not only NFκB studies, but studies of p53 and other dynamically controlled pathways. As such I have given keynote lectures, organized workshops, written invited reviews (72, 74, 78, 81), and edited a special issue devoted to dynamics in biological systems (71).
Our ongoing efforts are focused on (i) obtaining a higher resolution view of dynamics with live cell imaging studies, and we have developed experimental systems to examine primary cells from wild-type and knockout mice (previous studies employed overexpression of fluorescent reporters in established cell lines); (ii) understanding how NFκB dynamics are interpreted by target genes, and we have developed dynamic control mutants appropriate for transcriptomic studies, modeling approaches to interpret the responses, and tools for expression studies in single cells (RNA FISH and reporters); and (iii) strategies for manipulating dynamic control pharmacologically to achieve stimulus-specific inhibition of NFκB or inhibition of a subset of target gene expression. These efforts are supported by a multi-PI R01 (CA141722) grant on IKK (with G. Ghosh), a multi-PI R01 (GM089976) grant on single cells studies (with Hasty and Tsimring), a co-PI share of an R01 (GM072024) grant (with Levchenko), and a portion of a P01 (GM071862) grant (with Komives).
Stress responses determined by steady states associated with cell types and disease
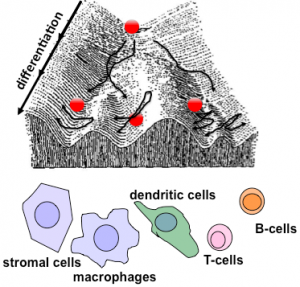 Using the multi-dimer NFκB signaling system as a model system, we have begun to study how stress response networks may be altered by environmental conditioning or priming (45, 54, 55, 74), or how they may change during differentiation to produce cell-type specific control. Iterative modeling and experimental studies showed that control of RelB by TLRs in dendritic cells is mediated by the canonical pathway and identified two key parameters that account for cell-type specific control, rather than a qualitatively cell type-specific molecular mechanism (88). Similar studies have characterized the dimer repertoire of mature B-cells and T-cells. To develop a predictive understanding, we extended our modeling efforts to account for NFκB dimer generation (49). These efforts uncovered a positive (“chaperone”) role of IκB proteins in NFκB control (Tsui et al.), confirming that the systematic and iterative analysis of the network will yield important new insights. We have developed a theoretical framework that allows us to probe the relationship between the cell type-specific steady state of a signaling system and its dynamic response to perturbation, not only computationally but analytically (82, 89).
Using the multi-dimer NFκB signaling system as a model system, we have begun to study how stress response networks may be altered by environmental conditioning or priming (45, 54, 55, 74), or how they may change during differentiation to produce cell-type specific control. Iterative modeling and experimental studies showed that control of RelB by TLRs in dendritic cells is mediated by the canonical pathway and identified two key parameters that account for cell-type specific control, rather than a qualitatively cell type-specific molecular mechanism (88). Similar studies have characterized the dimer repertoire of mature B-cells and T-cells. To develop a predictive understanding, we extended our modeling efforts to account for NFκB dimer generation (49). These efforts uncovered a positive (“chaperone”) role of IκB proteins in NFκB control (Tsui et al.), confirming that the systematic and iterative analysis of the network will yield important new insights. We have developed a theoretical framework that allows us to probe the relationship between the cell type-specific steady state of a signaling system and its dynamic response to perturbation, not only computationally but analytically (82, 89).
These studies are beginning to impact the direction of the field as indicated by our cover-art in the NFκB handbooks (66, 74) and the 2014 Keystone NFκB meeting organization.
Ongoing efforts are focused on (i) developing a predictive model of how all fifteen possible NFκB dimers are generated such that differentiation-associated network changes can be recapitulated and potentially manipulated; (ii) examine how the NFκB system is transformed during the differentiation of ES cells (which contain no NFκB) into neuronal and hematopoietic lineages (which contain distinct forms); and (iii) examining the mechanisms that may underlie NFκB misregulation during oncogenic transformation and cellular senescence. These efforts are supported by a P01 (GM071862) project (with Komives), an R01 (AI083453) on B-cells, and a pending stem cell (ES and iPS) CIRM grant.
Innate Immune Responses: Coordination of several signaling systems
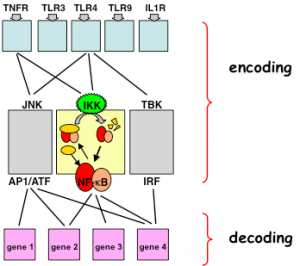 Within the innate immune responses the NFκB system is coordinated with the IRF and the MAPK signaling systems. Through iterative system modeling and experimentation we have begun to develop a predictive understanding of IRF activation and NFκB control via canonical and non-canonical pathways (88), and via Myd88 and TRIF. Initial studies revealed unexpected cross-regulation of a subset of IRF response elements by the NFκB family member p50:p50 (76). We are now developing tools to examine NFκB and IRF signaling in single cells, and a modeling framework to examine the functional consequence of coordinated signaling at the level of gene expression (Cheng et al, Rios et al).
Within the innate immune responses the NFκB system is coordinated with the IRF and the MAPK signaling systems. Through iterative system modeling and experimentation we have begun to develop a predictive understanding of IRF activation and NFκB control via canonical and non-canonical pathways (88), and via Myd88 and TRIF. Initial studies revealed unexpected cross-regulation of a subset of IRF response elements by the NFκB family member p50:p50 (76). We are now developing tools to examine NFκB and IRF signaling in single cells, and a modeling framework to examine the functional consequence of coordinated signaling at the level of gene expression (Cheng et al, Rios et al).
Ongoing efforts are focused on developing a predictive understanding of (i) TLR/RLR-specific control of NFκB and IRF transcription factors; (ii) the control and function of autocrine/paracrine cytokine mechanism on NFκB; (iii) the control and function of autocrine/ paracrine IFNγ on IRF/ISGF3 control. These efforts are supported by an R01 (GM071573) on TLR to NFκB signaling, a portion of a P50 (GM085764) grant (with Chanda), and a portion of the P01 (AI090935-01) on HIV (PI Young).
Systems Biology of the Nucleus: Integrating processes to predict gene expression
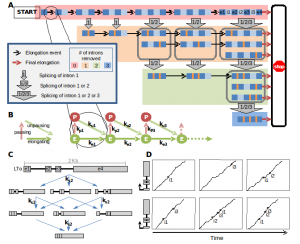 Using the innate immune gene expression response as a model system we are developing expertise to predict gene expression via a series of interconnected models that predict (i) transcription factor binding as a function of the DNA sequence/PWMs, the chromatin landscape, and TF-TF interactions (Hoff et al), (ii) promoter activity as a function of chromatin interaction data, (iii) elongation and splicing control (Davis-Turak et al), and (iv) mRNA export and half-life (Cheng et al). Leveraging my graduate training in the biochemistry of transcription and my recent expertise in modeling, these efforts are nascent and are supported by a portion of the P50 (GM085764) grant (with Ren, Glass, Johnson) and, potentially, a pending Keck award.
Using the innate immune gene expression response as a model system we are developing expertise to predict gene expression via a series of interconnected models that predict (i) transcription factor binding as a function of the DNA sequence/PWMs, the chromatin landscape, and TF-TF interactions (Hoff et al), (ii) promoter activity as a function of chromatin interaction data, (iii) elongation and splicing control (Davis-Turak et al), and (iv) mRNA export and half-life (Cheng et al). Leveraging my graduate training in the biochemistry of transcription and my recent expertise in modeling, these efforts are nascent and are supported by a portion of the P50 (GM085764) grant (with Ren, Glass, Johnson) and, potentially, a pending Keck award.
Host-Pathogen Interactions: Multi-scale modeling of immune responses
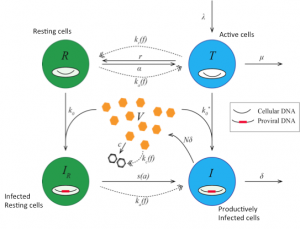 Coordinated pathogen-responsive signaling and gene expression results in a cellular immune effector response. We have begun to study both innate adaptive immune responses with experimental and computational approaches. Supported by a consortium of researchers we have developed a model for early HIV infection events within cells (Cheng et al). In our own laboratory we have studied B- and T-cell proliferation (Alves et al, Almaden et al) and developed computational tool to interpret CFSE data (Shokhirev et al) and genetic imprinting in mice (84).
Coordinated pathogen-responsive signaling and gene expression results in a cellular immune effector response. We have begun to study both innate adaptive immune responses with experimental and computational approaches. Supported by a consortium of researchers we have developed a model for early HIV infection events within cells (Cheng et al). In our own laboratory we have studied B- and T-cell proliferation (Alves et al, Almaden et al) and developed computational tool to interpret CFSE data (Shokhirev et al) and genetic imprinting in mice (84).
Ongoing efforts are focused on (i) extending the model to the multi-cellular compartment of the mucosa to study the role of cell-cell interactions and cytokines in conjunction with ex vivo cell culture systems; (ii) develop multi-scale models of lymphocyte proliferation such that intra-cellular molecular signaling networks can be probed for their function in determining the population dynamics of lymphocytes. These efforts are supported by an R01 (AI083453) on B-cells and a portion of the P01 (AI090935-01) on HIV (PI Young).
